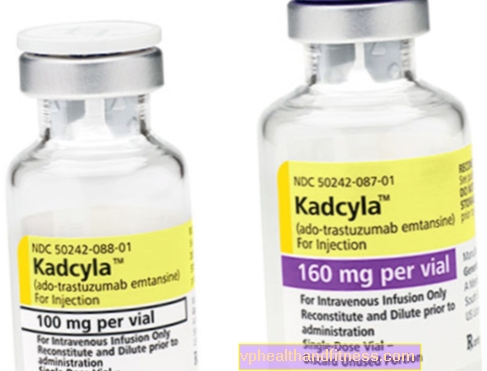
1 flakon sulandırıldıktan sonra infüzyon için 20 mg / ml konsantrasyonda 5 ml (8 ml) trastuzumab emtansine konsantre solüsyonundan 100 mg (160 mg) toz içerir.
| İsim | Paket içeriği | Aktif madde | Fiyat% 100 | Son düzenleme |
| Kadcyla | 1 şişe, hazırlık için toz son çözüm inf için. | Trastuzumab kostansin | 2019-04-05 |
Aksiyon
Antikanser ilaç. Trastuzumab emtansin, stabil bir MCC tiyoeter bağlantısı (4- sikloheksan-1-karboksilat) aracılığıyla DM1'e (maytansin türevi, mikrotübül inhibitörü) kovalent olarak bağlanan insanlaştırılmış bir anti-HER2 IgG1 monoklonal antikoru olan trastuzumab içeren bir antikor-ilaç konjugatıdır. . Emtansin, MCC-DM1 kompleksidir. DM1'in trastuzumab ile konjugasyonu, HER2 aşırı eksprese eden tümör hücrelerine karşı sitotoksik ilaçların seçici etkisine neden olur, böylece DM1'in hücre içi konsantrasyonunu doğrudan tümör hücrelerinde arttırır. HER2'ye bağlandıktan sonra trastuzumab emtansin, reseptör aracılı içselleştirmeyi takiben lizozomal degradasyon olup, DM1 içeren katabolitleri (esas olarak lizin-MCC-DM1) salar. Trastuzumab emtansinin etki mekanizmasına trastuzumab ve DM1'in aktivitesi aracılık eder. Trastuzumab emtansin, trastuzumab gibi, reseptörün hücre dışı alanının (ECD) IV. Bölgesine ve ayrıca Fcy reseptörlerine ve tamamlayıcı C1q'ya bağlanır. Ayrıca, HER2 reseptörünün ECD alanının aktivitesini inhibe eder, fosfatidilinozitol 3-kinaz (PI3-K) yolağının sinyalleşmesini inhibe eder ve HER2'yi aşırı eksprese eden insan meme kanseri hücrelerinde antikora bağımlı hücresel sitotoksisiteye (ADCC) aracılık eder. DM1, tubuline bağlanır. Tübülin polimerizasyonunu inhibe ederek, hem DM1 hem de trastuzumab emtansin, hücrelerin hücre döngüsünün G2 / M fazında tutuklanmasına ve sonuçta apoptoz yoluyla hücre ölümüne yol açmasına neden olur. MCC bağlayıcı, sistemik DM1 salımını azaltır ve hedef bölgedeki konsantrasyonunu artırır. Trastuzumab emtansin dekonjuge edilir ve daha sonra hücre lizozomlarında proteoliz ile katabolize edilir. DM1, esas olarak CYP3A4 tarafından ve daha az ölçüde de CYP3A5 tarafından metabolize edilir. Trastuzumabın T0.5'i yaklaşık 4 gündür. 3 haftalık aralıklarla verilen çoklu intravenöz infüzyonları takiben trastuzumab emtansin birikimi olmamıştır. Yaşın, trastuzumab kostansinin farmakokinetiği üzerinde etkisi yoktur.
Dozaj
Damardan. Hazırlık bir doktor tarafından yazılmalı ve kanser hastalarının tedavisinde deneyimli sağlık uzmanlarının gözetimi altında uygulanmalıdır. Trastuzumab emtansin alan hastalarda HER2-pozitif kanser - immünohistokimya (IHC) skoru 3+ veya in situ hibridizasyonda (ISH) oran ≥2 olmalıdır. Testler, CE işaretli in vitro diagnostik (IVD) testleri kullanılarak yapılmalıdır. CE IVD testi mevcut değilse, test başka bir doğrulanmış test ile yapılmalıdır. Tıbbi hatalardan kaçınmak için, hazırlanan ve uygulanan ilacın Herceptin (trastuzumab) değil Kadcyla (trastuzumab emtansine) olduğundan emin olmak için flakon etiketini kontrol etmek önemlidir. Önerilen doz 3,6 mg / kg vücut ağırlığıdır. 3 haftada bir intravenöz infüzyon olarak verilir (21 günlük döngü). Hastalar, tümör progresyonuna veya kabul edilemez toksisite sağlanana kadar tedavi edilmelidir. Başlangıç dozu, 90 dakikalık intravenöz infüzyon şeklinde uygulanmalıdır. İlaç, uygulama sırasında deri altından geçebileceğinden enjeksiyon bölgesi yakından izlenmelidir. Önceki infüzyon iyi tolere edilmişse, sonraki dozlar 30 dakikalık infüzyon olarak verilebilir. Hastalar infüzyon sırasında ve sonrasında en az 30 dakika gözlenmelidir. Hasta infüzyonla ilişkili semptomlar geliştirirse infüzyon hızı yavaşlatılmalı veya kesilmelidir. Hayati tehlike arz eden infüzyon reaksiyonları durumunda trastuzumab emtansin kesilmelidir. Acil kullanım için alerjik / anafilaktik infüzyon reaksiyonları için ilaçlar ve acil durum ekipmanı hazır bulundurulmalıdır. Planlanmış bir doz atlanırsa, mümkün olan en kısa sürede uygulanmalıdır; bir sonraki döngüye kadar beklemeyin. Dozlama programı, 3 haftalık doz aralığını koruyacak şekilde ayarlanmalıdır. Bir sonraki doz, dozlama önerilerine göre uygulanmalıdır. Semptomatik yan etkilerin yönetimi, tedavinin periyodik olarak kesilmesini, dozun azaltılmasını veya tedavinin kesilmesini içerebilir. Doz azaltma çizelgesi (başlangıç dozu 3.6 mg / kg): ilk doz azaltımı 3 mg / kg; ikinci doz azaltımı 2.4 mg / kg canlı ağırlık; Daha fazla doz azaltılması gerekirse, tedavi durdurulmalıdır. AST ve ALT yükselmeleri için doz modifikasyon yönergeleri: Derece 2 (> 2,5 ila ≤5 x NÜS) - doz ayarlaması gerekmez; 3. derece (> 5 ila ≤20 x ULN) - AST / ALT, derece ≤ 2'ye (> 2,5 ila 20 x NÜS) düşerse preparatı kullanın - tedaviyi sonlandırın. Hiperbilirubinemi için doz değiştirme kuralları: 2. derece (> 1.5 ila ≤3 x ULN) - preparatı bilirubin seviyesi ≤1'e düştüğünde kullanın. (> ULN ila 1.5 x ULN), doz ayarlaması gerekli değildir; 3. derece (> 3 ila ≤10 x ULN) - bilirubin konsantrasyonu ≤ derece 1'e (> ULN ila 1.5 x ULN) düşerse preparatı kullanın, ardından dozu azaltın (bkz. doz azaltma çizelgesi); Derece 4 (> 10 x ULN) - tedaviyi sonlandırın. Trombositopeni için doz değiştirme kuralları: 3. derece (trombosit konsantrasyonu 25.000 ila 3) - preparatı trombosit konsantrasyonu ≤1 olduğunda kullanın. (örneğin, ≥75.000 / mm3); doz ayarlaması gerekmez; Derece 4 (trombosit konsantrasyonu 3) - preparatı, trombosit konsantrasyonu ≤1'e ulaştığında kullanın. (örn. 75.000 / mm3) daha sonra dozu azaltın (bkz. doz azaltma programı). Ventriküler disfonksiyonlu hastalarda doz modifikasyon ilkeleri: LVEF% 45 - tedaviye devam edin; LVEF% 40 ila% 45 ejeksiyon fraksiyonunun aynı anda azaltılması ile - preparatı kullanmayın; LVEF'i 3 hafta içinde tekrar değerlendirin, LVEF hala başlangıca göre 10 yüzde puanı ise, tedaviyi bırakın; semptomatik KKY - tedaviyi durdurun. Çocuklar ve ergenler: Bu popülasyonda yaygın meme kanseri (MBC) bilinmediğinden, 18 yaşın altındaki çocuklarda ve adolesanlarda güvenlik ve etkililik belirlenmemiştir. Özel hasta grupları. Derece 3 veya 4 periferik nöropati gelişen hastalarda, Derece 2'ye ulaşana kadar tedavi geçici olarak kesilmelidir; Tedaviye yeniden başlarken, doz azaltma çizelgesinde özetlendiği gibi dozun azaltılması düşünülebilir. 65 yaş ve üstü hastalar için doz ayarlamasına gerek yoktur; 75 yaş ve üzerindeki hastalarda tedavinin güvenliğini ve etkinliğini belirlemek için yeterli veri yoktur. Hafif veya orta derecede böbrek yetmezliği olan hastalarda başlangıç dozu ayarlamasına gerek yoktur; Şiddetli böbrek yetmezliği olan hastalarda potansiyel doz ayarlaması ihtiyacı belirlenemez ve bu nedenle bu hastalar dikkatle izlenmelidir. Hafif veya orta derecede karaciğer yetmezliği olan hastalarda başlangıç dozu ayarlamasına gerek yoktur. Şiddetli karaciğer yetmezliği olan hastalarda trastuzumab emtansin çalışılmamıştır; Tedavi sırasında gözlenen bilinen hepatotoksisite nedeniyle karaciğer yetmezliği olan hastalar tedavi edilirken dikkatli olunmalıdır. Verme yolu. Hazırlık tıbbi personel tarafından çözülmeli ve seyreltilmeli ve intravenöz infüzyon olarak uygulanmalıdır. Bolus veya hızlı enjeksiyon olarak verilmemelidir.
Belirteçler
Daha önce trastuzumab ve bir taksanla kombinasyon halinde veya ayrı ayrı tedavi edilmiş, HER2 pozitif, ameliyat edilemez lokal olarak ilerlemiş veya metastatik meme kanseri olan yetişkin hastaların monoterapisi. Lokal olarak ilerlemiş veya genelleşmiş hastalık için önceden tedavi görmüş veya adjuvan tedaviyi tamamlama sırasında veya tamamlandıktan sonraki 6 ay içinde relaps gösteren hastalar
Kontrendikasyonlar
Etkin maddeye veya preparatın diğer bileşenlerine aşırı duyarlılık.
Önlemler
Trastuzumab emtansin ile yapılan klinik çalışmalarda pnömoni dahil interstisyel akciğer hastalığı (ILD) vakaları bildirilmiştir; bazıları akut solunum yetmezliği ile ilişkilendirilmiştir veya ölümcül olmuştur. İAH veya pnömoni teşhisi konulan hastalarda preparatla tedavinin kesilmesi önerilir. İlerlemiş kanser ve ilişkili hastalıkların komplikasyonları ile ilişkili istirahatte dispnesi olan hastalar, pulmoner komplikasyonlar geliştirme riski yüksek olabilir. Preparat ile tedavi sırasında, karaciğerde nodüler rejeneratif hipertrofi (NRH) ve ilaca bağlı karaciğer hasarına bağlı ölümler dahil olmak üzere karaciğer ve safra yolunda ciddi bozukluklar ve hepatotoksisite gözlendi; Hepatotoksik potansiyele sahip olduğu bilinen komorbiditeler ve / veya eşzamanlı ilaçlar da etkilenmiş olabilir. Tedaviye başlamadan önce ve sonraki dozlamadan önce karaciğer fonksiyonu izlenmelidir. Başlangıçta ALT yükselmeleri olan hastalar (örn. Karaciğer metastazları ile ilişkili), karaciğer yetmezliği gelişimine yatkındır ve Derece 3-5 hepatik toksisite veya yüksek karaciğer laboratuar seviyeleri riski altındadır. NRH vakaları, karaciğer biyopsileri alınarak teşhis edildi. NRH gelişimi klinik portal hipertansiyon belirtileri ve / veya karaciğerin BT taramasında görülen siroz benzeri bir tablo olan, ancak normal serum transaminaz seviyeleri olan ve siroz olduğuna dair başka bir kanıt bulunmayan tüm hastalarda dikkate alınmalıdır. NRH teşhisi konulursa, preparatla tedavi kesilmelidir. Tedaviye başlamadan önce serum transaminazları> 2.5 x ULN (normalin üst sınırı) veya total bilirubin> 1.5 x ULN olan hastalarda çalışılmamıştır. Serum transaminaz aktivitesi> 3 x ULN ve aynı zamanda toplam bilirubin konsantrasyonu> 2 x ULN olması durumunda tedavi sonlandırılmalıdır. Karaciğer yetmezliği olan hastaların tedavisinde dikkatli olunmalıdır. Tedavi sırasında sol ventrikül disfonksiyonu riski artar. KADCYLA ile tedavi edilen hastalarda, 50 yılda sol ventrikül ejeksiyon fraksiyonunda (LVEF) bir azalma, başlangıçta düşük bir LVEF değeri (25 kg / m2) ile gözlenmiştir. Standart kardiyak fonksiyon testleri (ekokardiyogram veya çok kapılı radyoizotop anjiyografi) tedaviye başlamadan önce ve düzenli aralıklarla (örn. Her 3 ayda bir) yapılmalıdır. Hastaların LVEF'si başlangıçtaki çoğu klinik çalışmada ≥% 50'dir. Randomizasyondan önceki 6 ay içinde konjestif kalp yetmezliği, tedavi gerektiren ciddi aritmiler, miyokardiyal enfarktüs öyküsü veya kararsız anjina veya ilerlemiş kanser nedeniyle istirahat sırasında dispnesi olan hastalar çalışma dışı bırakıldı. Sol ventrikül bozuklukları durumunda, sonraki doz ertelenmeli veya tedavi sonlandırılmalıdır. İnfüzyonla ilişkili bir reaksiyon nedeniyle trastuzumab tedavisini bırakan hastalarda trastuzumab tedavisinin etkisi araştırılmamıştır; Bu hasta grubunda bu ilaçla tedavi önerilmemektedir. İnfüzyonla ilişkili şiddetli reaksiyon gelişen hastalarda, semptomlar düzelene kadar tedaviye ara verilmelidir. Tedavinin yeniden başlatılması, reaksiyonun ciddiyetine ilişkin klinik yargıya dayalı olarak düşünülmelidir. Hayatı tehdit eden bir infüzyon reaksiyonu durumunda tedavi kesilmelidir. Trastuzumab tedavisinin, aşırı duyarlılık nedeniyle trastuzumab tedavisi kesilen hastalarda etkisi araştırılmamıştır; Bu hastalarda trastuzumab emtansin kullanımı önerilmemektedir. Hastalar hipersensitivite / alerjik reaksiyonlar için izlenmelidir; bu semptomlar infüzyonla ilişkili reaksiyonlarla aynı olabilir; şiddetli anafilaktik reaksiyonlar gözlemlenmiştir. Aşırı duyarlılık (sonraki infüzyonlar sırasında artan reaksiyonlarla) gelişirse, trastuzumab KADCYLA tedavisi kesilmelidir.Hemorajik olay riskinden dolayı (CNS, solunum ve gastrointestinal hemoraji dahil), antikoagülanlar veya antiplatelet ilaçlarla birlikte uygulandığında dikkatli olunmalı ve ek izleme düşünülmelidir. Her trastuzumab emtansin dozunun uygulanmasından önce trombosit sayımlarının izlenmesi önerilir. Trombositopenili hastalar (≤100.000 / mm3) ve antikoagülan tedavi alan hastalar (örn. Varfarin, heparin, düşük molekül ağırlıklı heparinler) trastuzumab ile tedavi sırasında yakından izlenmelidir. Trastuzumab emtansin, tedaviye başlamadan önce trombosit sayısı ≤100.000 / mm3 olan hastalarda çalışılmamıştır. Trombositopeninin Derece 3 veya daha yüksek (3) kötüleşmesi durumunda preparat, toksisite Derece 1'e (75.000 / mm3) düşene kadar kullanılmamalıdır. KADCYLA ile yapılan klinik çalışmalarda, esas olarak duyusal olmak üzere derece 1 periferik nöropati meydana gelmiştir. Derece 3 periferik nöropatisi olan hastalar, klinik araştırmalara katılımdan çıkarılmıştır. Derece 3 veya 4 periferik nöropatili hastaların tedavisi, komplikasyon Derece 2'ye düşene kadar periyodik olarak kesilmelidir. Hastalar, nörotoksisite belirtileri açısından düzenli olarak izlenmelidir. Biyolojik preparatların izlenebilirliğini iyileştirmek için, uygulanan ilacın ticari adı hasta dosyasına açıkça yazılmalıdır (veya belirtilmelidir). Bu popülasyonda yaygın meme kanseri bulunmadığından, 18 yaşın altındaki çocuklarda ve ergenlerde ilacın güvenliliği ve etkililiği belirlenmemiştir. Bolus veya hızlı enjeksiyon olarak verilmemelidir.
İstenmeyen aktivite
Çok yaygın: idrar yolu enfeksiyonu, trombositopeni, anemi, hipokalemi, uykusuzluk, periferik nöropati, baş ağrısı, kanama, burun kanaması, öksürük, dispne, stomatit, ishal, kusma, bulantı, kabızlık, ağız kuruluğu, karın ağrısı , döküntü, kas-iskelet ağrısı, artralji, miyalji, yorgunluk, ateş, asteni, titreme, transaminazlarda artış. Yaygın: nötropeni, lökopeni, aşırı duyarlılık, baş dönmesi, disguzi, hafıza bozukluğu, kuru göz sendromu, konjunktivit, görme bozuklukları, gözyaşı artışı, sol ventrikül disfonksiyonu, hipertansiyon, dispepsi, dişeti kanaması, cilt kaşıntısı, alopesi, tırnak hastalığı , palmar-plantar eritrodisestezi, ürtiker, periferik ödem, alkalin fosfatazda artış, infüzyonla ilişkili reaksiyonlar (ciltte kızarıklık, titreme, ateş, dispne, düşük tansiyon, hırıltı, bronkospazm, taşikardi). Yaygın olmayan: pnömoni (ILD), hepatotoksisite, karaciğer yetmezliği, nodüler rejeneratif hiperplazi, portal hipertansiyon, enjeksiyon yeri ekstravazasyonu (eritem, hassasiyet, cilt tahrişi, ağrı veya şişme). Ek olarak, hiperbilirubinemi gözlenmiştir. Klinik çalışmalarda, trastuzumab emtansin ile tedavi sırasında serum transaminazlarında (Derece 1-4) yükselmeler meydana geldi ve genellikle geçiciydi. Transaminaz yükselmesi çoğunlukla geçiciydi ve dozlamadan sonraki 8. günde zirve yaptı. Daha sonra, toksisite Derece 1'e düştü veya bir sonraki döngüden önce çözüldü. Ayrıca kümülatif bir etki de vardı (Derece 1-2 ALT / AST yükselmesi olan hastaların yüzdesi sonraki sikluslarla arttı). Transaminaz yükselmiş hastalarda, hastaların çoğu, son trastuzumab emtansin dozundan sonraki 30 gün içinde 1. derece toksisitede azalma veya iyileşme yaşamıştır. Klinik araştırmalara katılan hastaların% 2.2'sinde sol ventrikül disfonksiyonu bulundu. Çoğu vakada, Derece 1 veya 2 LVEF düşüşleri asemptomatikti.Genellikle tedavinin ilk döngüleri sırasında, hastaların% 0.4'ünde Derece 3 veya 4 toksisite gözlendi (1-2). Tüm hastaların% 2.2'sinde ciddi hemorajik olay epizotları (Derece ≥3) bildirilmiştir. Klinik çalışmalarda hastaların% 24.9'unda trombositopeni meydana geldi ve tedavinin kesilmesine yol açan en yaygın advers reaksiyondu. Klinik çalışmalarda, Asyalı hastalarda trombositopeni insidansı ve şiddeti daha yüksekti. Bu komplikasyonları geliştiren bazı hastalar ayrıca antikoagülan tedavi ile tedavi edildi. Anti-trastuzumab emtansin antikorları için pozitif test edilen hastaların% 5.3'ünde CNS kanaması dahil olmak üzere ölümcül kanama atakları ve ciddi kanama komplikasyonları vardı.
Gebelik ve emzirme
Gebe kadınlarda trastuzumab emtansin kullanımı önerilmemektedir. Hamile kalmadan önce, kadınlar fetüse zarar verme olasılığı konusunda bilgilendirilmelidir. Ancak hamile kalan hastaların hemen bir doktora başvurması gerekir. Gebe bir kadın trastuzumab emtansin ile tedavi edilirse, multidisipliner bir ekip tarafından yakın takip önerilir. Trastuzumab hamile bir kadına uygulandığında fetal hasara veya ölüme neden olabilir. İlaçla tedavi edilen hamile kadınların çocuklarında, bazıları ölümcül akciğer hipoplazisine yol açan oligohidramnios vakaları bildirilmiştir. Trastuzumab emtansinin sitotoksik bileşeni olan DM1 teratojenik ve potansiyel olarak embriyotoksik olabilir. Trastuzumab KADCYLA tedavisine başlamadan önce kadınlar emzirmeyi bırakmalıdır. Hastalar tedaviyi bıraktıktan 7 ay sonra emzirmeye başlayabilir. Çocuk doğurma potansiyeli olan kadınlar, ilaçla tedavi sırasında ve son trastuzumab KADCYLA dozunu takip eden 7 ay boyunca etkili bir doğum kontrolü kullanmalıdır. Etkili doğum kontrol yöntemleri erkekler veya kadın partnerleri tarafından da kullanılmalıdır.
Yorumlar
İnfüzyonla ilişkili reaksiyonlar yaşayan hastalar, bu semptomlar çözülene kadar araç veya makine kullanmamalıdır. Esas alınarak hazırlanan bilgiler SPC 13 Temmuz 2017 Mevcut SmPC www.roche.pl adresinde mevcuttur.
Etkileşimler
İnsan karaciğer mikrozomlarındaki metabolizma çalışmalarının in vitro sonuçları, DM1'in esas olarak CYP3A4 enzimi tarafından ve daha az ölçüde de CYP3A5 tarafından metabolize edildiğini göstermektedir. Güçlü CYP3A4 inhibitörlerinin (örn. Ketokonazol, itrakonazol, klaritromisin, atazanavir, indinavir, nefazodon, nelfinavir, ritonavir, sakinavir, telitromisin ve vorikonazol) birlikte kullanımından kaçınılmalıdır çünkü bu, DM1 düzeylerini ve toksisiteyi artırabilir. CYP3A4'ü inhibe etmeyen veya çok az inhibe eden alternatif bir formülasyon düşünülmelidir. Güçlü CYP3A4 inhibitörlerinin eşzamanlı kullanımından kaçınılamıyorsa ve mümkünse, trastuzumab emtansin uygulamasını CYP3A4 inhibitörleri dolaşımdan temizlenene kadar (yaklaşık 3 inhibitörün yarılanma ömrü) ertelemeyi düşünün. Bununla birlikte, güçlü bir CYP3A4 inhibitörü eşzamanlı olarak kullanılıyorsa ve trastuzumab emtansin ile tedavi geciktirilemezse, bu gibi durumlarda hasta yan etkiler açısından yakından izlenmelidir.
Hazırlık şu maddeyi içerir: Trastuzumab emtansine
Geri ödenen ilaç: HAYIR